What causes Sickle Cell Disease?
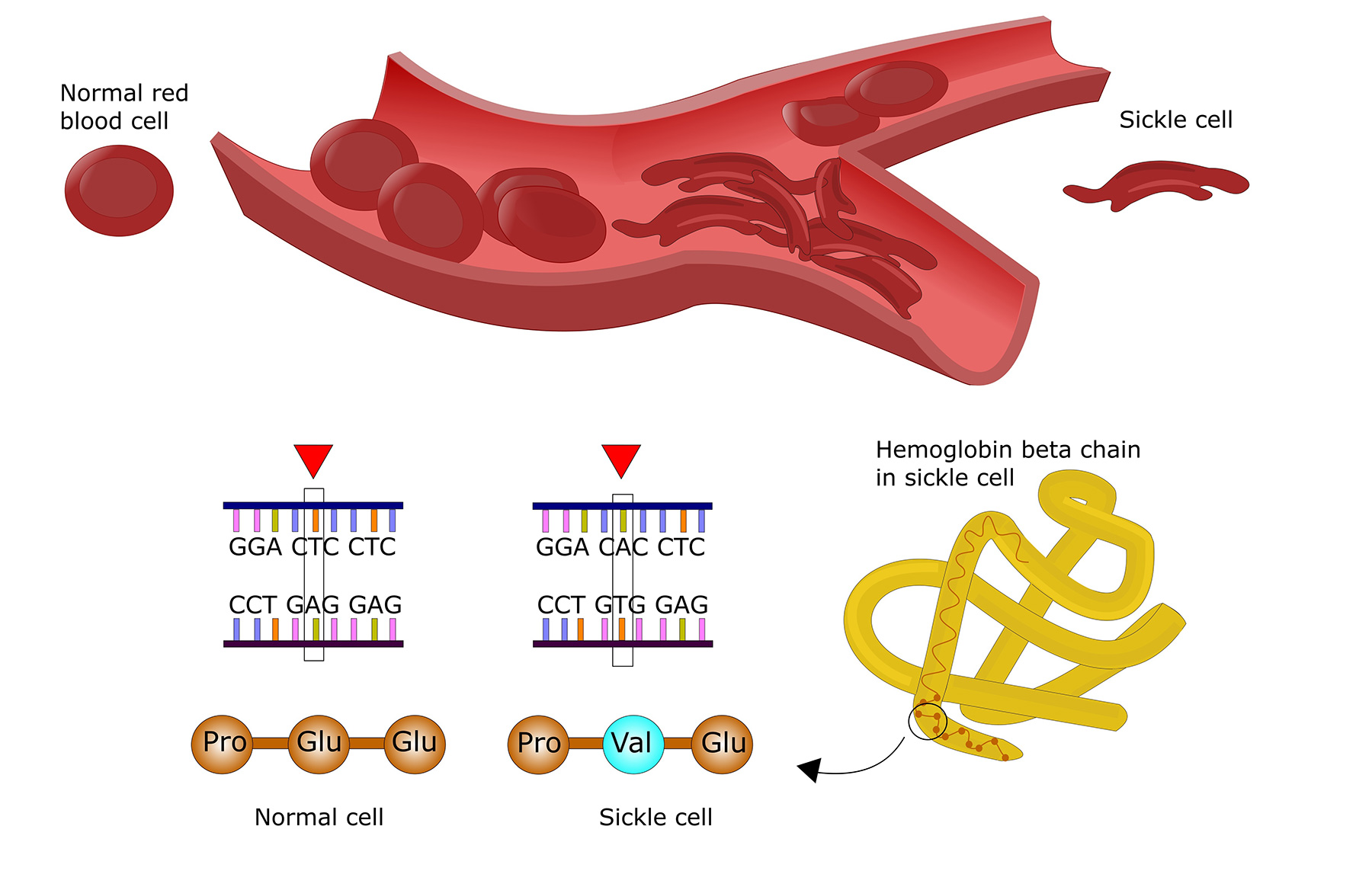
Hemoglobin is the protein in red blood cells that carries oxygen to the rest of the body. Normal hemoglobin (HbA) is made up of four protein subunits: two α-globin chains and two β-globin chains.
Sickle cell disease (SCD) is caused by genetic mutations in the HBB gene. HBB encodes the hemoglobin β-globin subunit. Different genetic mutations in the HBB gene produce different variants of abnormal hemoglobin (S, C or E):
HbS – The 20A>T mutation in the HBB gene results in the production of hemoglobin S (HbS)
HbC – The 19G>A mutation in the HBB gene results in the production of hemoglobin C (HbC)
HbE– The 79G>A mutation in the HBB gene results in the production of hemoglobin E (HbE)
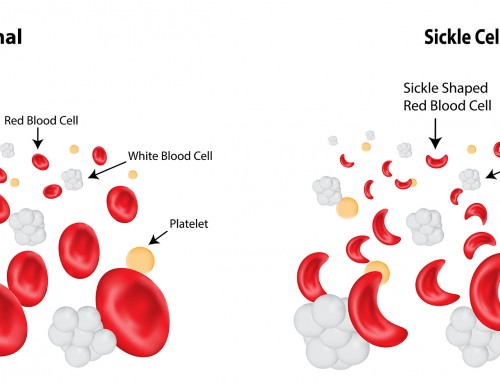
Abnormal hemoglobin is stiff, sticky and distorts the red blood cell into a sickled shape. These abnormal red blood cells have a shorter lifespan as they die prematurely, leading to anemia. They can also stick to blood vessels and cause blockage or slow down oxygen and blood supply throughout the body.
As SCD is an autosomal recessive disease, two defective copies of the HBB gene must be inherited to develop this disorder. People affected with sickle cell disease have at least one copy of the HbS variant of the hemoglobin β-globin subunit. In the most severe form of sickle cell disease, also known as sickle cell anemia, two copies of HbS are inherited. In other types of sickle cell disease, one copy of HbS and a copy of a different abnormal β-globin subunit are inherited. A common type of SCD is hemoglobin SC disease, where one copy of HbS and HbC each are inherited. Hemoglobin SE disease is a rare type of SCD, and it occurs when one copy of HbS and one copy of HbE are inherited.
People who have only one defective copy of the abnormal hemoglobin, with another normal copy of hemoglobin are referred as silent carriers, and they typically do not show signs and symptoms of SCD as they can make enough normal hemoglobin (HbA). However, if two carriers have children, then their children are at risk of developing sickle cell disease.
References:
Bender MA, Douthitt Seibel G (2003, updated 2014 Oct 23). Sickle Cell Disease. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.
About SCD & SCT. Sickle Cell Disease Association of America. Accessed March 2016.
Schnog JB, Duits AJ, Muskiet FAJ, ten Cate H, Rojer RA, Brandjes DPM (2004). Sickle cell disease; a general overview. The Netherlands Journal of Medicine. 62(10): 364-374.
DNA In the News2017-04-06T20:55:08+00:00