What is Sickle Cell Disease?
Sickle cell disease is caused by genetic defects that result in the production of abnormal hemoglobin
Find out if you are at risk - get tested
| Test Type | Testing Time | Fee |
All orders received before 3pm PST / 6pm EST are shipped out the same business day. All orders received after 3pm PST / 6pm EST or on weekends or holidays are shipped out the following business day. 24/7 online status check and account management available for all tests.
Sickle cell diseases are a group of inherited blood disorders characterized by the production of abnormal hemoglobin.
Hemoglobin
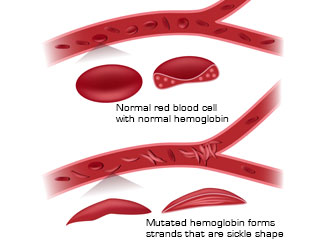
Hemoglobin is a protein component of red blood cells that carries oxygen from the lungs to the rest of the body. The most common hemoglobin in a normal adult is hemoglobin A (HbA), composed of two α-globin chains and two β-globin chains. Several hundred mutations have been identified in the genes that encode α-globin and β-globin. Mutations in the HBA1 and HBA2 genes (encode α-globin) lead to alpha thalassemia disorders. Mutations in the HBB gene (encodes β-globin) result in several different disorders, including beta thalassemias, methemoglobinemia and sickle cell disorders. Each of these mutations results in the production of abnormal hemoglobin, affecting the function of this important molecule and often changing the number and shape of the affected person’s red blood cells.
Normal red blood cells are disc shaped, a shape that makes them flexible and allows them to easily travel through the blood vessels to deliver oxygen. The abnormal hemoglobin found in people with the sickle cell disorder is stickier than normal and forms stiff rods within the red blood cells. These rods change the shape of red blood cells and give them the characteristic crescent or sickle shape. These sickled cells are not as flexible as normal red blood cells and can stick to blood vessels, causing blockages or slowing blood flow. As a result, oxygen is not delivered effectively to the rest of the body, contributing to the disease symptoms. Sickling can be enhanced by conditions such as low oxygen levels, dehydration and increased acidity of the blood.
What Causes Sickle Cell Disease?
The characteristic sickle shape of red blood cells in sickle cell disorders occurs due to the presence of the HbS form of hemoglobin. Sickle cell disease is generally an autosomal recessive disease, which means two defective copies of HBB must be inherited to develop the symptoms of the disease. However, even those that carry just one defective HbS alelle (known as the sickle cell trait), produce some abnormal hemoglobin and can experience symptoms under extreme conditions. The severity of sickle cell disease depends on how many copies of HbS are inherited. Individuals with two copies of the HbS mutation, have sickle cell anemia (or HbSS disease), which accounts for 60%-70% of the sickle cell cases in the US. Sickle cell compound heterozygotes occur when a person inherits two different abnormal alleles – one HbS allele and one other defective HBB allele. Hemoglobin SC disease (HbSC) occurs in people with one HbS and one HbC allele and has varying severity, but is generally less severe than HbSS disease. Another form, known as Hemoglobin SE disease (HbSE), occurs in people with one HbS and one HbE allele, and is associated with milder symptoms. It is also possible to inherit an HbS allele and an HBB gene carrying a beta thalassemia mutation, leading to sickle/beta thalassemia disease. This disease also has varying severity, depending on the form of the beta thalassemia mutation.
How Common is Sickle Cell Disease?
Sickle cell disease is one of the more common blood disorders in the US. It is more common in individuals with African, South Asian, Southern European, Middle Eastern and South and Central American ancestry. There are about 90,000 to 100,000 people with sickle cell disease in the US. It is less common in the UK and France with 10,000 – 15,000 affected people. The prevalence of sickle cell disease is highest in sub-Saharan Africa. Sickle cell disease occurs in about 1 in 500 African-Americans and 1 in 36,000 Hispanic-American births.
It is estimated that 300 million people around the world carry just one HbS allele and have the sickle cell trait. The high prevalence of this disorder occurs due to the malaria resistance that defective HBB genes provide. Hence the HbS allele is more common in areas of high malaria incidence, particularly in West Africa (1 in 4 people carry an HbS allele) and those of Afro-Caribbean descent (1 in 10). It also occurs in approximately 1 in 12 African-Americans, 1 in 150 individuals of European descent, 1 in 50 Asians and 1 in 100 Northern Greeks.
Next, Symptoms of Sickle Cell Disease »
About SCD & SCT. Sickle Cell Disease Association of America. Accessed March 2016.