Symptoms of Sickle Cell Disease
Symptoms are due to the decreased ability to effectively deliver oxygen around the body
Find out if you are at risk - get tested
| Test Type | Testing Time | Fee |
All orders received before 3pm PST / 6pm EST are shipped out the same business day. All orders received after 3pm PST / 6pm EST or on weekends or holidays are shipped out the following business day. 24/7 online status check and account management available for all tests.
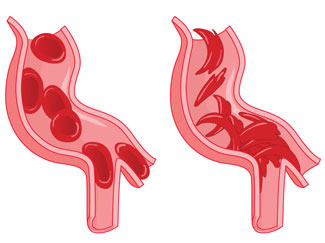
Sickling of Red Blood Cells
The most distinguishing feature of sickle cell disorder is sickle shaped red blood cells (RBCs). Most of the symptoms are caused by these sickled RBCs and the resulting inability to effectively deliver oxygen around the body. The characteristic sickle shape occurs due to the clumping of abnormal hemoglobin molecules into stiff rods, hence changing the shape and reducing the flexibility of the RBC.
Chronic hemolysis
Chronic hemolysis is the rupture of RBCs, and is one of the hallmark characteristics of sickle cell disease. The sickled RBCs lack the flexibility of normal RBCs, and they burst open more frequently. This reduces the lifetime of these sickled RBCs to only last 10 – 20 days, compared to normal RBCs that are only replaced every 120 days. This premature breakdown and the short lifespan of RBCs is what causes anemia – the reduced number of RBCs. Anemia is associated with a variety of symptoms, including fatigue, weakness, delayed growth and jaundice.
Intermittent Vaso-occlusive Episodes
A second characteristic of sickle cell disease includes intermittent vaso-occlusive episodes. These occur when sickled RBCs block blood vessels and restrict blood and oxygen supply to tissues and organs, resulting in severe tissue damage. Certain organs that are predisposed to lower oxygen levels or acidity include the spleen, liver and kidneys. These organs are more prone to tissue damage, because lower oxygen levels and acidity both enhance sickling of the RBCs. Organs that have high metabolism rates, such as the brain and muscles, tend to extract more oxygen from the blood and also enhance sickling.
Pain Crises
Pain crises or recurrent painful episodes are another result of vaso-occlusive episodes. Pain is caused by sickled blood cells traveling through the tiny blood vessels in the chest, abdomen or joints. Pain also occurs in the bones and can vary in intensity and duration (lasting from a few hours to a few weeks).
Hand-Foot Syndrome
Hand-foot syndrome is when the hands and feet swell due to the sickled RBCs reducing circulation, and is one of the first symptoms that manifest in infants. Infants with sickle cell disease have no symptoms at birth, because their predominant form of hemoglobin is fetal hemoglobin. Fetal hemoglobin does not contain any β-globin and is instead composed of two α-globin chains and two γ-globin chains. As the fetal hemoglobin is replaced with adult hemoglobin (two α-globin and two β-globin chains) the affects of the defective HBB gene (β-globin) become apparent. Signs of sickle cell anemia will manifest by the age of four months and affected children develop life-threatening anemia and jaundice and they fail to thrive. Adolescents with sickle cell disease will also experience delayed puberty.
Other complications
Other complications can arise from sickle cell disease, including vision problems from vaso-occlusions damaging the retina, frequent infections due to damage in the spleen, high blood pressure in the blood vessels that supply the lungs (which can lead to heart failure), and gallstones from the increased breakdown of RBCs.
In cases of undiagnosed sickle cell disease, if any of the following symptoms appear you should seek medical attention immediately: unexplained episodes of pain, swelling of hands and/or feet, abdominal swelling, fever, pale or yellow-tinted skin and any symptoms of a stroke.
Next, Diagnosis of Sickle Cell Disease »
About SCD & SCT. Sickle Cell Disease Association of America. Accessed March 2016.